Designing primers that work well is essential for successful molecular biology experiments like PCR and qPCR. First, you need to grasp the basics of primers; they are short sequences that must be complementary to your target DNA. Good primers usually range from 18-24 bases in length, with a GC content between 40-60% and can ideally have a melting temperature (Tm) of 50-60°C. Next, gather the necessary sequence information from databases like NCBI. Use tools such as Primer-BLAST or Primer3 to design your primers while keeping in mind factors like amplicon size and specificity. Afterward, validate them through in silico analysis and PCR testing to confirm their effectiveness before troubleshooting any possible issues during experiments. Happy experimenting!
1. Understand Primer Basics
 Credits: en.wikipedia.org
Credits: en.wikipedia.org
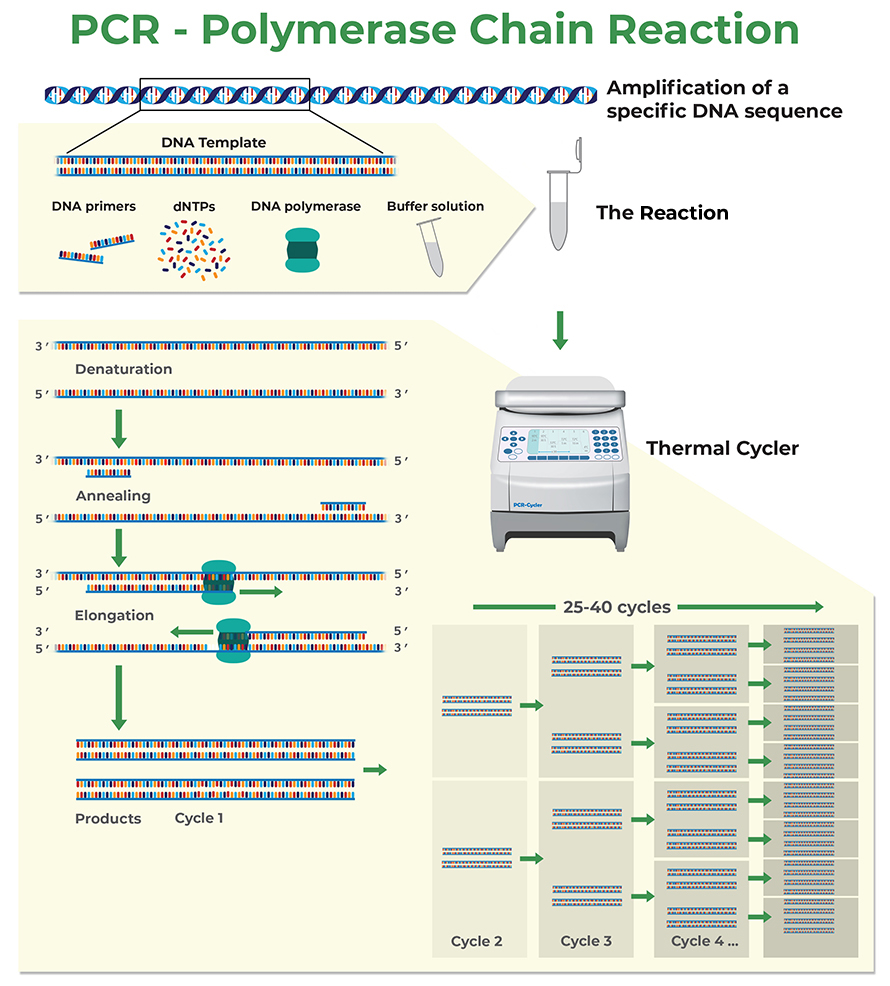
Primers are short sequences of nucleotides, usually 18-24 bases long, that serve as starting points for DNA synthesis in techniques like PCR. They must be complementary to the target DNA sequence to ensure specific binding and successful amplification. Good primers have certain key properties. Firstly, they should have a balanced GC content, ideally between 40-60%. This range is important because GC pairs form three hydrogen bonds, making them more stable than AT pairs, which only form two. Secondly, the melting temperature (Tm) of the primers is crucial; a Tm of 50-60°C is optimal, and the Tm of paired primers should be within 2-5°C of each other. This balance helps maintain consistent binding conditions during the PCR process. For example, if one primer has a Tm of 55°C, the other should ideally be between 53-57°C. By understanding these primer basics, you can set a solid foundation for designing effective primers for your experiments.
2. Gather Sequence Information
To design effective primers, you first need the nucleotide sequence of the target DNA. This information is crucial as it serves as the template for your primers to bind. Start by searching databases like NCBI or Ensembl to find the sequence of the gene or region you are interested in. For example, if you are studying a specific gene involved in cancer, you can enter its name in the NCBI database to retrieve its sequence. Once you have this sequence, you can use it in various primer design tools.
Additionally, consider the context of your experiment. If you are targeting a specific variant or mutation, ensure that the sequence you gather accurately reflects this. If the gene has multiple isoforms or alleles, you may need to select the most relevant sequence for your study. This careful selection will help ensure that your primers are specific and effective in amplifying the desired target.
3. Design the Primers
To design effective primers, start by using a reliable software tool like Primer-BLAST or Primer3. Input your target sequence and set specific parameters that suit your experiment’s needs. For example, if you are targeting a gene for qPCR, you might set your amplicon size to be around 100 bp, which is optimal for quantitative analysis. It’s also important to ensure that your primers have a balanced GC content, ideally between 40-60%, to promote stable binding during PCR.
Next, after generating a list of potential primers, evaluate them carefully. Look for primers that end with a G or C at the 3′ end, as this can significantly enhance the stability of the primer-template interaction. Additionally, check for any sequence homology to non-target regions to avoid unwanted amplification. For instance, if your target gene is involved in a pathway, ensure that the primers do not amplify any pseudogenes that may be present.
Consider designing a pair of primers with a Tm within 2-5°C of each other to ensure they anneal at similar temperatures during PCR. This uniformity helps in achieving consistent amplification results. If you’re working with mRNA, remember to design primers that span exon-exon junctions. This prevents amplifying any contaminating genomic DNA, providing cleaner results.
After you have chosen your primers, take the time to analyze them for potential issues such as self-dimerization or hairpin formation. These structural issues can interfere with the PCR process, leading to low yields or non-specific products. Tools like OligoCalc can help check for these problems. The more thorough your design process, the more likely your experiments will be successful.
| Step | Action | Details |
|---|---|---|
| 1 | Input the Sequence | Enter the nucleotide sequence of your target gene into Primer-BLAST. |
| 2 | Set Parameters | – Amplicon Size: Set the desired amplicon size (typically between 70-200 bp for qPCR). – Exon-Intron Selection: Ensure primers span exon-exon junctions. – Tm and GC Content: Specify desired Tm and GC content. |
| 3 | Analyze the Results | – Check the 3′ end of each primer for G or C residue. – Ensure no significant self-complementarity or dimer formation potential. |
4. Validate the Primers
 Credits: zymoresearch.com
Credits: zymoresearch.com
Once you have selected potential primer pairs, it is crucial to validate them to ensure they will work effectively in your experiments. Start with in silico analysis by using tools like Primer-BLAST to check for specificity. This step helps confirm that your primers will bind only to the intended target sequence and not to any non-target regions in the genome. Look for potential off-target binding sites that may lead to non-specific amplification.
After conducting the in silico analysis, the next step is to test the primers in a wet lab setting. Order the primers and perform a PCR experiment using your target DNA template. After running the PCR, utilize gel electrophoresis to separate the PCR products. Analyze the gel to confirm that you observe a single, specific band at the expected size. This indicates that your primers are functioning correctly and amplifying the desired target.
For instance, if your target is a gene of interest with a known size of 150 bp, a successful PCR should yield a band around that size on the gel. If you see multiple bands, it may indicate non-specific binding. It’s also essential to use high-quality template DNA to minimize the presence of inhibitors that could affect the PCR reaction.
Additionally, optimizing the PCR conditions is vital. Adjusting the annealing temperature based on the melting temperature (Tm) of your primers can improve specificity and yield. By carefully validating your primers, you increase the likelihood of success in your molecular biology experiments.
5. Troubleshooting Common Issues
During your experiments, you may face various challenges that can affect the success of your PCR reactions. Here are some common issues and troubleshooting tips to help you resolve them.
If you encounter *non-specific bands* on your gel, this often indicates that the primers are binding to unintended sites on the DNA template. To address this, you can try redesigning your primers to increase their specificity. Additionally, adjusting the annealing temperature can help; increasing the temperature can improve specificity by reducing the chance of non-specific binding.
In cases where you see *no amplification* at all, first ensure that your template DNA is of high quality. Contaminants or degraded DNA can hinder the reaction. Also, double-check your PCR protocol, especially the concentrations of primers, nucleotides, and the enzyme. Sometimes, simply optimizing the PCR conditions, such as the annealing temperature or extension time, can lead to successful amplification.
If you are getting multiple bands or a smear on your gel, this might indicate that the PCR reaction is not optimized. Try diluting your template DNA, as too much starting material can cause non-specific amplification. You might also need to adjust the number of cycles in your PCR, as too many cycles can lead to non-specific products.
For primer-dimer formation, which can often show up as unexpected bands, check the primer sequences for complementary regions that may cause them to bind to each other. If you find that your primers have a high potential for dimer formation, redesign them to minimize self-complementarity.
Lastly, always keep an eye on your *reaction controls*. Including positive and negative controls in your experiments can help you identify issues with your reagents or protocol. If the positive control works but your experimental samples do not, it may indicate a problem with your template or primer design.
- Check for nonspecific amplification
- Review primer concentrations
- Validate the template quality
- Assess thermal cycling conditions
- Consider primer-dimer formation
- Re-evaluate primer design parameters
- Ensure correct annealing temperatures
- Verify the presence of inhibitors
6. Additional Considerations for qPCR
When designing primers for qPCR, it’s important to consider the length of your amplicons. Keeping them between 70-140 base pairs is ideal for achieving efficient amplification and accurate quantification. Additionally, if you are using TaqMan probes for probe-based assays, ensure that the melting temperature (Tm) of the probes is higher than that of your primers. This will help maintain specificity during amplification.
Another key factor is the choice of the template. For qPCR, using cDNA synthesized from mRNA is common, especially when measuring gene expression levels. It’s advisable to design primers that span exon-exon junctions to prevent amplification of contaminating genomic DNA.
Also, consider the use of internal controls or reference genes to normalize your data and improve the reliability of your results. These controls should have similar amplification efficiencies to your target genes.
Finally, be aware of the potential effects of PCR inhibitors that may be present in your samples. This can include substances from the sample extraction process. Ensuring high-quality template preparation can significantly enhance your qPCR results.
7. Tips for Bisulfite PCR
 Credits: star-protocols.cell.com
Credits: star-protocols.cell.com
When designing primers for bisulfite PCR, it’s important to consider the unique challenges posed by bisulfite treatment of DNA, which converts unmethylated cytosines into uracils. This means that your primers must be designed to accommodate these changes. First, aim for primer lengths between 26 to 30 bases. This length is effective in ensuring specificity and helps in amplifying the converted DNA. Second, avoid including CpG sites in your primer sequences, as these are crucial for distinguishing between methylated and unmethylated states. Including them can lead to non-specific binding and reduced amplification efficiency.
For example, if your target sequence has a methylated CpG site, design your primers to flank this region without including the CpG itself. Additionally, ensure the 3′ ends of your primers are targeted towards the converted cytosines, which will now be thymidine in the PCR product. Lastly, validate your primers using a methylated DNA template to confirm their effectiveness before proceeding with experiments.
8. Final Checks for Primer Design
Before you finalize your primer design, conduct a thorough review to ensure the best possible performance in your experiments. First, confirm that both primers are free of significant self-complementarity, which can lead to unintended dimer formation. Use tools like the IDT OligoAnalyzer to check the primer sequences for any potential hairpin structures or dimers.
Next, assess the specificity of the primers against the entire genome. This step is crucial, especially if you are working with complex genomes. You want to make sure your primers only amplify the target sequence and do not bind to other regions. Use databases like NCBI or UCSC Genome Browser for this analysis.
Also, consider the presence of single nucleotide polymorphisms (SNPs) within your target region. SNPs can affect primer binding and may lead to variations in amplification efficiency. If your target region has known SNPs, redesign the primers accordingly to avoid these polymorphic sites.
Finally, double-check the final sequences for any errors or ambiguities. Any small mistake in the primer sequence can lead to failed experiments. Once satisfied with the design, you can proceed to the ordering and testing phase.
Frequently Asked Questions
1. What are primers in experiments and why are they important?
Primers are short pieces of DNA that help start the process of copying DNA during experiments. They are important because they ensure that the right part of the DNA is copied accurately.
2. How do I know the right length for my primers?
The ideal length for primers is usually between 18 to 25 nucleotides. This helps to ensure they bind properly to the DNA without being too specific or too loose.
3. What should I consider when choosing the melting temperature (Tm) for my primers?
When choosing the melting temperature, aim for a Tm of about 50-65 degrees Celsius. It’s important that the Tm of both primers in a pair is similar to ensure they work well together.
4. Can I use software tools to design my primers?
Yes, there are many software tools available that can help you design primers by calculating the optimal length, Tm, and specific binding sites based on your DNA sequence.
5. What is the purpose of testing my primers after designing them?
Testing your primers is crucial to confirm they work as expected, meaning they bind only to the target DNA and produce clear results without unwanted extra products.
TL;DR This guide provides clear steps for designing effective primers for PCR and qPCR experiments. Start by understanding primer basics, such as length, GC content, and melting temperature. Gather the necessary sequence information from databases like NCBI, then use tools like Primer-BLAST or Primer3 to design primers based on your target gene. Validate your primers through in silico analysis and PCR testing to ensure specificity. Troubleshoot common issues like non-specific amplification and low yield, and consider specific guidelines for qPCR and bisulfite PCR. Finally, check for potential SNPs and avoid structural issues in your primer sequences.